四毛&八毛 微算云平台 2024年10月01日 08:18 广东
成果介绍
同时活化金属和晶格氧位点,通过提供高可用性的活性位点和介导催化活性/稳定性来构建兼容的多机制催化剂,有望用于析氧反应(OER),但仍存在重大挑战。
武汉理工大学木士春教授等人在OER过程中对NiMoO4·xH2O@Fe,S进行完全重构,得到Fe和S双调制的NiFe氢氧化物(R-NiFeOOH@SO4),并通过原位光谱/质谱和化学探针证实,在金属/氧位点同时优化的情况下,实现了相容的吸附质演化机制和晶格氧氧化机制。进一步的理论分析表明,Fe促进了吸附质演化机制下的OER动力学,而S激发了晶格氧氧化机制下的晶格氧活性,表现为O 2p能带中心上移、d-d库仑相互作用放大、金属-氧键减弱、中间吸附自由能优化。
得益于兼容的多机制,R-NiFeOOH@SO4只需要251±5/291±1 mV的过电位就可以在碱性介质中获得100/500 mA cm-2的电流密度,并且具有超过300小时的稳定性能。这项工作为更好地设计高性能OER催化剂提供了理解OER机理的见解。
相关工作以《Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation》为题在《Nature Communications》上发表论文。
值得注意的是,该工作受到了三位审稿人的一致认可!无论是所提出的新概念,还是论文所提供的证据、结论都能够清晰有力地令人信服。

众所周知,吸附质演化机制(AEM)和晶格氧氧化机制(LOM)是常见的OER机制,目前更多的研究主要集中在如何操纵催化剂活性位点以改善、促进这两种机制,改善催化性能。该研究论文创新地提出了AEM与LOM相容于一个催化剂上,获得了惊人的效果!
图文导读
图1 催化剂的制备与结构
通过化学蚀刻和原位电化学自重构工艺合成了SO42-修饰的NiFe氢氧化物(R-NiFeOOH@SO4),如图1a所示。首先,通过水热反应在泡沫镍(NF)衬底上生长NiMoO4水合物(NiMoO4?xH2O)纳米棒(图1b),然后将其作为模板在化学蚀刻过程中引入Fe和S物质。得到的表面粗糙的NiMoO4?xH2O@Fe,S预催化剂仍主要表现为NiMoO4?xH2O晶体结构,结晶度减弱(图1c)。然后,通过阳极循环伏安法(CV)活化合成R-NiFeOOH@SO4,去除NiMoO4?xH2O自牺牲模板。据此,通过NiMoO4?xH2O和NiMoO4?xH2O@Fe阳极活化得到R-NiOOH和R-NiFeOOH作为对照样品。
FESEM和TEM图像显示,所有催化剂都继承了初始的纳米棒结构(图1d、e),R-NiFeOOH@SO4在表面表现出明显的颗粒细化。活化后,R-NiFeOOH@SO4的BET表面积增加。HRTEM图像(图1f)揭示了NiMoO4?xH2O纳米线的高结晶度,而NiMoO4?xH2O@Fe,S(图1g)和NiMoO4?xH2O@Fe存在明显的表面非晶化和更多的晶格缺陷,进一步证实了蚀刻导致的结晶度下降,这更有利于OER重构。在阳极CV活化后,HRTEM图像中几乎看不到属于初始NiMoO4?xH2O的晶格间距(图1h),晶格间距0.208和0.240 nm分别对应于NiOOH(210)和(011)晶面,确定了NiMoO4?xH2O模板成功转化为NiOOH活性物。此外,EDS(图1i)证实了所有催化剂中Mo元素几乎完全去除,而Ni、Fe、S和O元素均匀分布在R-NiFeOOH@SO4的整个纳米棒区域。
图2 催化剂的电子结构与配位结构
本研究利用XPS和XAS进一步分析了催化剂的电子结构和配位结构。相对于NiMoO4?xH2O, NiMoO4?xH2O@Fe和NiMoO4?xH2O@Fe,S的Ni 2p能谱显著地向较低的结合能偏移(图2a),同时Mo的3d结合能也向较高的结合能偏移,表明电子从Mo迁往Ni位点。CV活化后,所有催化剂的Ni 2p结合能都出现了异常的负位移,这应该是由于在非原位测试下,不稳定的NiOOH转化为Ni(OH)2。但NiMoO4?xH2O@Fe、S和R-NiFeOOH@SO4之间的负位移较小(-0.24 eV),且R-NiFeOOH@SO4的Ni氧化态高于R-NiOOH和R-NiFeOOH,说明Ni2+的转化率较低,这是由于其更有利的完全重构过程。
特别是,在O 1s谱(图2b)中,衍生化氢氧化氧的金属-晶格氧(M-O)信号表现出明显的正偏移,这表明晶格氧(M-O(2-δ)-)的氧化参与了电化学活化过程。其中,R-NiFeOOH@SO4的M-O偏移更为显著(0.4 eV)。同时,与NiMoO4?xH2O@Fe,S相比,R-NiFeOOH@SO4的氧空位含量由于OH-在电解液中的再填充而降低。此外,R-NiFeOOH@SO4的Fe 2p信号峰在图2c中向更高的结合能移动,表明表面Fe物质向更高的氧化态转变,而R-NiFeOOH的Fe 2p结合能保持稳定在+3价。如图2d所示,R-NiFeOOH@SO4的S 2p信号完全转化为氧化S物种,对应于硫酸盐(SO42-)的特征信号峰。
Fe、S的引入对NiMoO4?xH2O的Ni位点电子结构的调整可以通过Ni的K边XANES光谱进一步证实。CV活化后,所有催化剂的吸收边都向低能方向移动,其中R-NiFeOOH@SO4的移动程度较低,与R-NiOOH和R-NiFeOOH相比,位于能量最高的位置(图2e),与XPS分析结果一致。与NiMoO4?xH2O@Fe相比,Fe的K边XANES(图2f)在NiMoO4?xH2O@Fe,S中也显示出较低的平均Fe氧化态,并在R-NiFeOOH@SO4中向高能量转移。此外,S的K边XANES光谱证明R-NiFeOOH@SO4中的S物种以SO42-的形式存在,进一步揭示了S物种的氧化。 图3 电催化OER性能
图3 电催化OER性能
在1 M KOH中,最优R-NiFeOOH@SO4只需要251±5 mV的OER过电位就可以实现100 mA cm-2的电流密度,低于R-NiOOH(367±4 mV)、NiFe-LDH(286±6 mV)和R-NiFeOOH(284±7 mV)(图3a、b)。特别是R-NiFeOOH@SO4可以在291±1和308±1 mV的过电位下获得500和1000 mA cm-2的高电流密度。如图3c所示,它还具有最低的Tafel斜率(56 mV dec-1)和最小的电荷转移电阻(Rct),表明其具有良好的OER动力学和高效的电荷转移。
另一方面,对于R-NiFeOOH@SO4(图3d),在3000次循环CV扫描前后均未观察到明显的衰减,甚至在100和500 mA cm-2下稳定运行300 h,仅略有衰减(图3e),充分说明了OER催化稳定性的显著性。经过长期稳定性测试,通过SEM、TEM、EDX、拉曼和XPS表征,R-NiFeOOH@SO4的纳米珊瑚状结构和组成仍然保持良好,证明了其坚固的结构框架。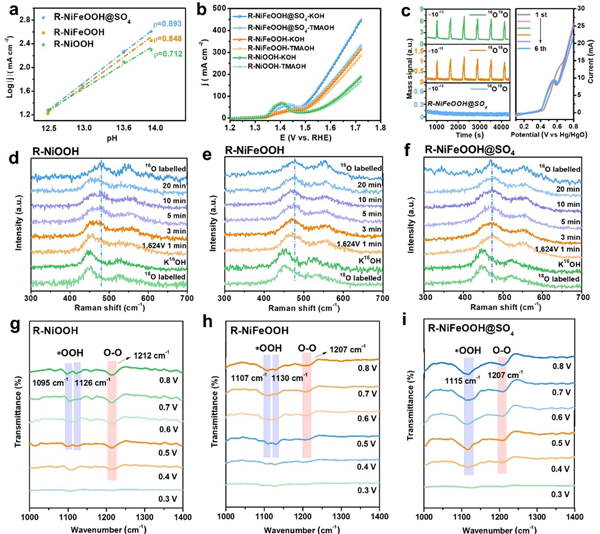
图4 兼容AEM-LOM的OER催化分析
本研究进一步探讨了R-NiFeOOH@SO4参与的协同催化机制和OER活性的来源。在图4a中,所有催化剂的OER活性都表现出一定程度的pH依赖性,而R-NiFeOOH@SO4的电流密度随着pH值的降低而下降得更快,这说明晶格氧参与了OER过程。此外,R-NiFeOOH@SO4在1 M TMAOH中表现出明显的OER活性减弱,这是由于在四烷基铵阳离子(TMA+)的强结合下抑制了LOM,而它对R-NiFeOOH和R-NiOOH只有轻微的活性衰减(图4b)。
为了进一步验证催化剂晶格氧在OER过程中的参与,采用了原位18O同位素标记微分电化学质谱(DEMS)方法。催化剂被标记18O,以进一步检测在含有16O的电解质中OER过程中O2的释放。如图4c所示,所有18O标记催化剂的DEMS测量结果均呈现m/z=32和m/z=34的信号,表明16O2和16O18O气体的生成,证实了有一个晶格氧参与了释放O2。未经18O标记的R-NiFeOOH@SO4的DEMS结果只显示16O16O信号,没有检测到明显的16O18O。同时,与R-NiOOH相比,R-NiFeOOH的16O18O峰面积增大,而其16O18O/16O16O的面积比减小,说明在AEM途径下,Fe调制对析氧的贡献更为显著。
此外,用18O同位素标记的原位拉曼光谱也证实了晶格氧参与了OER过程。如图4d-f所示,在氧质量对振动模式的影响下,18O标记的催化剂明显向低波数偏移。在含16O的电解液中,在1.624 V恒定电位下,随着18O的消耗,18O标记催化剂的拉曼峰逐渐移回常规16O标记催化剂的位置,其中R-NiFeOOH@SO4晶格氧释放更快,拉曼峰在1 min内移回。相比之下,R-NiFeOOH和R-NiOOH需要20 min甚至更长时间才能释放标记的18O。这一结果进一步证实了R-NiFeOOH@SO4在OER过程中更快的晶格氧释放,揭示了S物种调制对晶格氧的有效活化。
图5 DFT计算
为了更深入地了解OER协同机理与活度优化之间的联系,进一步进行了DFT计算。选择实际OER活性物质NiOOH、NiFeOOH和NiFeOOH@SO4作为计算模型。首先,计算O 2p轨道的态密度,以反映图5a中的晶格氧活度。与NiOOH(-2.773 eV)和NiFeOOH(-2.814 eV)相比,NiFeOOH@SO4的O 2p能带中心(-2.626 eV)相对于费米能级(EF)呈上升趋势,有利于氧位点电子的移除,揭示了S调制对晶格氧的有效活化。
如图5b所示,计算得到NiOOH、NiFeOOH和NiFeOOH@SO4的UHB/LHB中心位置分别为1.05/-2.5、1.12/-2.7和1.14/-2.86 eV。NiFeOOH@SO4(4 eV)的d-d库仑相互作用(U)值大于NiFeOOH(3.82 eV)和NiOOH(3.55 eV),进一步证实了其有利于晶格氧活化。通过计算COHP来评价金属-氧键的强度。在图5c中,NiOOH、NiFeOOH和NiFeOOH@SO4的量化Ni-O键强度分别为0.752、0.711和0.666。NiFeOOH@SO4较低的-ICOHP值表明Fe,S共调制诱导更多的电子被填入反键轨道,导致Ni-O键变弱,助长了晶格氧的参与,并在LOM途径中形成氧空位。
在AEM和LOM途径下,计算了NiOOH、NiFeOOH和NiFeOOH@SO4的吉布斯吸附自由能图。在AEM途径中分别考虑了Ni原子和Fe原子作为吸附位点的吸附自由能。如图5d所示,在Fe,S共改性下,NiFeOOH@SO4模型在Ni位点上的RDS能垒优化为1.92 eV。引入Fe后,*OH在Fe位点的去质子能势垒(RSD)急剧下降,NiFeOOH和NiFeOOH@SO4的去质子能势垒分别为1.66 eV和1.64 eV,这意味着活性位点转向了Fe(图5e),这从理论上解释了Fe引入后OER活性的大幅增强。
LOM的吉布斯自由能图如图5f所示,确定*O-OH的形成为NiOOH、NiFeOOH和NiFeOOH@SO4的RDS,其能垒分别为2.06、3.18和2.01 eV。计算结果表明,单一Fe的引入不利于LOM途径,这可能与Fe位点的竞争AEM热力学有关,而S物种(SO4)的调制有效地优化了LOM途径中的OER活性。因此,Fe、S共调制实现了AEM和LOM途径下对NiFeOOH@SO4的OER中间体吸附的同时优化,缩小了AEM和LOM之间的能垒差异,提高了多机制的相容性(图5g, h)。文献信息
Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation,Nature Communications,2024.
https://www.nature.com/articles/s41467-024-52682-y